Test Name
Chromosomal Microarray SNP, Constitutional (CRMSNP)
CPT Code
81229
Methodology
Genomic Oligonucleotide-SNP Microarray
Turnaround Time
10 – 14 days
Specimen Requirements
Type:
Whole blood
Volume:
4 mL
Minimum Volume:
1 mL
Specimen Container:
Lavender BD Hemogard™ K2EDTA Tube
Transport Temperature:
Ambient
Stability
Ambient:
48 hours
Refrigerated:
72 hours
Frozen:
Unacceptable
Reference Range
Refer to report
Additional Information
Background Information
Evidence-based approaches, coupled with technological advancements in the field of genetics, are changing how the human genome is analyzed and interpreted for diagnostic purposes. Chromosomal microarray analysis (CMA) is a recommended first-tier test for diagnosing unexplained intellectual disabilities, dysmorphic features, congenital anomalies, and autism.1-5 This technique compares relative fluorescent signal intensities of two different genomes, a test, and a reference, that compete for hybridization to DNA sequences representing the whole human genome in order to detect a gain or loss of genetic material. However, CMA does not detect allelic imbalances resulting in absence of heterozygosity (AOH).
Advances in the field of genetics and diagnostic medicine have resulted in the refinement of the existing CMA platform by incorporating single nucleotide polymorphism (SNP) markers into the existing non-polymorphic markers used to detect gains and losses of DNA. The analysis of SNP data provides information about the allelic imbalances associated with AOH. Identifying regions of excessive homozygosity on a single chromosome could suggest uniparental disomy (UPD), which may warrant further clinical and laboratory investigation when observed on chromosomes with known imprinting disorders associated with UPD. In addition, the detection of excessive homozygosity on multiple chromosomes may suggest consanguinity and, therefore, could be useful in determining candidate genes for further testing for autosomal recessive disorders.
The Agilent array is comprised of 107,000 oligonucleotide 60mer probes spaced one probe every 40kb across the backbone of the array and one probe every 10kb in targeted clinically significant regions in the genome. For SNP analysis, the array features 60,000 SNP 60mer probes with an effective resolution down to 10Mb. Data for the SNP markers are displayed to demonstrate either homozygosity (AA or BB) or heterozygosity (AB) at every SNP locus. AOH is indicative of hemizygosity if the corresponding CMA data shows a deletion of the region. If the CMA data is normal, AOH signifies homozygosity.
Clinical Indications
In the pediatric population, many abnormal phenotypes are associated with chromosomal imbalances that can be identified using CMA to detect copy number change (CNC). Thus, whole-genome CMA has replaced chromosome and FISH studies to become the first-tier test for the evaluation of children with unexplained developmental disabilities, intellectual disabilities, dysmorphic features, congenital anomalies, and autism.
Based on numerous published studies, the yield of pathogenic or clinically significant CNC by CMA is approximately 15-20% in a pediatric population, compared with a yield of 3-5% by standard cytogenetic analysis in the same population. Variants of uncertain clinical significance (VOUS), or clinical significance unknown, are found in less than 10% and could play an important role in the clinical diagnosis. To a great extent, parental and family studies can be helpful in the clinical interpretation of these unknowns, as de novo occurrence of the CNC is more likely related to a pathogenic event.
CMA testing for CNC is recommended as a first-line test in the initial postnatal evaluation of individuals with the following:
- Multiple anomalies not specific to a well-delineated genetic syndrome
- Apparently nonsyndromic DD/ID
- Autism spectrum disorders
This is not a recommended test for adult patients with multiple miscarriages or pregnancy losses and no abnormal phenotype. Chromosome analysis is a better test to exclude the possibility of balanced rearrangements that cause pregnancy losses.
Appropriate follow-up is recommended in cases of chromosome imbalance identified by CMA and may include cytogenetic/FISH or other molecular studies of the patient or parents, and clinical genetics consultation. In addition to detecting CNC, the SNP microarray can also be used to diagnose suspected uniparental disomy (UPD) or imprinting disorder, the possible absence of heterozygosity to determine the degree of relatedness by identity-by-descent (autozygosity), and autosomal recessive condition risks. Additional testing may be necessary to confirm disorders associated with the absence of heterozygosity.
SNP testing may be used to identify:
- A chromosomal abnormality or micro-duplication/deletion syndrome with a normal karyotype
- The size of a duplication/deletion involved in an unbalanced translocation
- Triploidy
- Cryptic duplications/deletions in a phenotypically abnormal individual with an apparently balanced karyotype
- Uniparental disomy (UPD)
- Absence of heterozygosity to determine the degree of relatedness by identity-by-descent (autozygosity)
Methodology
The SNP test involves DNA extraction, restriction enzyme digestion, labeling, purification, hybridization, washing, array scanning, analysis, and interpretation. DNA extracted from the patient’s peripheral blood is digested, labeled, and hybridized to the microarray. Following hybridization, the microarray is scanned, and the signal intensities are collected then compared to a reference in order to determine copy number changes and absence of heterozygosity.
Whole-genome SNP microarray testing at Cleveland Clinic utilizes the GGXChip + SNP v1.0 platform, which contains non-polymorphic and polymorphic probes to detect both copy number changes (CNC) and allelic imbalances (SNP probes) within the same array. For CNC analysis, the array is comprised of 107,000 oligonucleotide 60mer probes spaced one probe every 40kb across the backbone of the array, and one probe every 10kb in targeted clinically-significant regions in the genome. For SNP analysis, the array features 60,000 SNP 60mer probes with an effective resolution down to 10Mb. In the backbone regions, the resolution for copy number detection is approximately 120kb and in the targeted regions it is approximately 30kb.
In our validation, the resolution for AOH was detected at approximately 1.5Mb, but the resolution is dependent on SNP probe coverage. For clinical purposes, AOH greater than 10Mb will be reported; however, in chromosomes associated with imprinting disorders, smaller changes will be evaluated further and may be reported. For a complete list of clinically-recognized regions of the genome and imprinted chromosomes please visit www.signaturegenomics.com.6
Interpretation
A written summary of the microarray test is provided in the Test Overview. Gains and losses are reported based on genomic content in line with ACMG guidelines for microarray interpretation. Copy number variations (CNV) or CNC devoid of relevant gene content or reported as common findings in the general population may not be reported.
A copy number change of uncertain clinical significance may be detected and will be reported per ACMG guidelines in one of three subcategories:3
- Uncertain clinical significance, likely pathogenic;
- Uncertain clinical significance, likely benign; or
- Uncertain clinical significance, no classification.
While most copy number changes observed by chromosomal microarray testing can readily be characterized as pathogenic or benign, there are limited data available to support the definitive classification of a subset into either of these categories. In these situations, a number of considerations are taken into account to help interpret results including the size and gene content of the imbalance, whether the change is a deletion or duplication, the inheritance pattern, and the clinical and/or developmental history of a transmitting parent. The continual discovery of novel copy number variations and published clinical reports means that the interpretation of any given copy number change may evolve with increased scientific understanding. The detection of excessive homozygosity may suggest the need for additional laboratory testing to confirm uniparental disomy or to test for mutations in genes associated with autosomal recessive disorders consistent with the patient’s clinical presentation that are present in regions of homozygosity.
Uniparental Disomy
Uniparental disomy (UPD), the presence of two copies of a chromosome or chromosomal region from a single parent, is clinically-relevant when it involves loci that undergo genomic imprinting.
There are two types of UPD:
- Isodisomy, in which the two parental copies are identical; and
- Heterodisomy, in which the parental copies are derived from both parental homologs and are, therefore, not identical.
SNP microarrays will detect isodisomy but not heterodisomy, since only isodisomy or segmental isodisomy results in homozygosity.
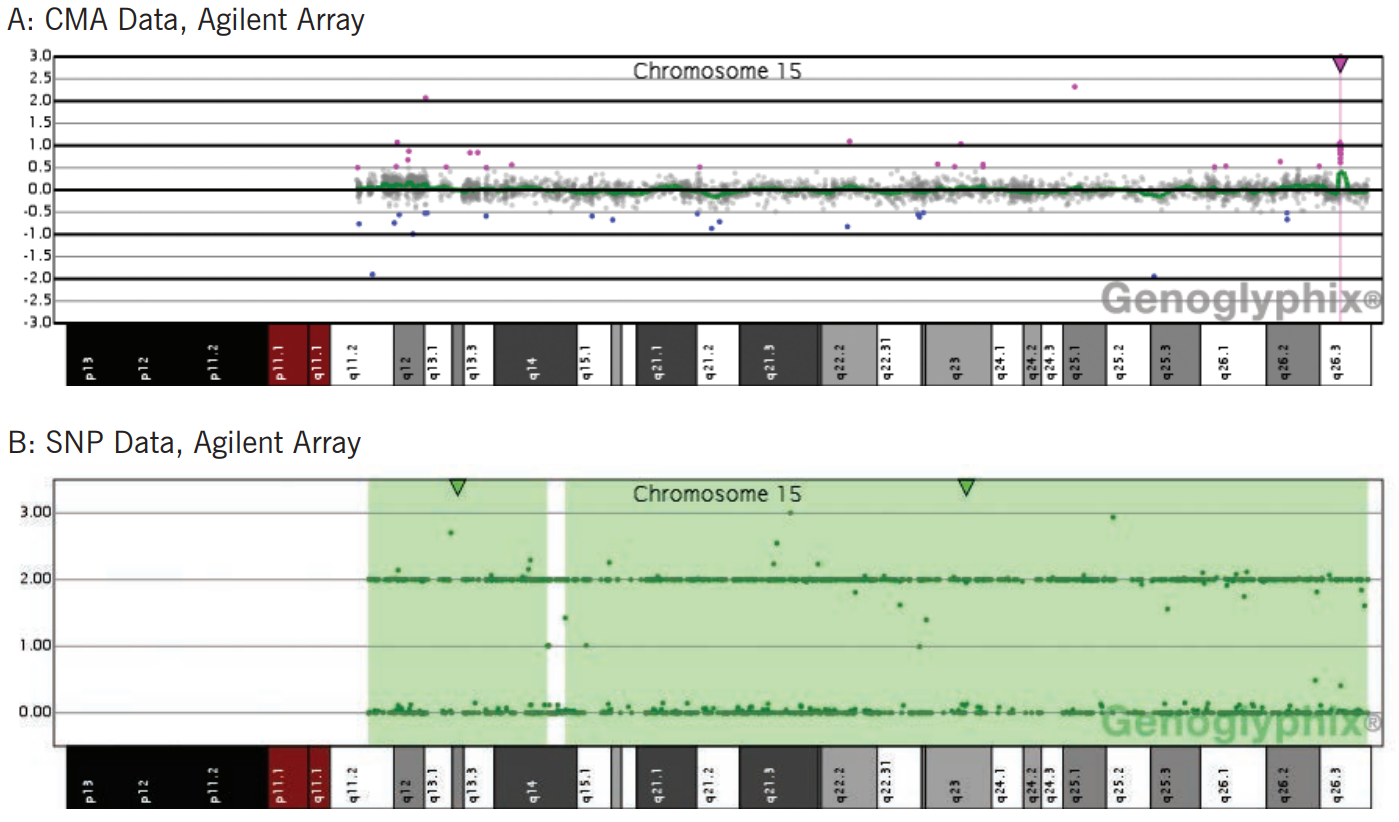
UPD is the most likely explanation for an absence of heterozygosity that is restricted to a single chromosome, especially if the region is very large. Several chromosome regions are imprinted and lead to an abnormal phenotype in the presence of UPD derived from a particular parent (Figure 1). UPD in other regions of the genome, even if covering a large region, is generally not considered to be a pathogenic finding in and of itself (see recessive disease discussion below).
Figure 1: Uniparental Disomy 15
Identity by Descent (IBD) and Consanguinity
The observation of multiple AOH regions, also known as long continuous stretches of homozygosity (LCSH), present on multiple chromosomes is generally assumed to reflect inheritance of these regions by descent from a common ancestor. This type of homozygosity is referred to as “identity by descent.” A single or a few small isolated stretches can be the result of a founder effect in an isolated population. The presence of especially long stretches on multiple chromosomes suggests the possibility of a more direct biological relationship between the parents (i.e., parental consanguinity) (Figure 2). A consanguineous relationship refers to the sharing of a common ancestor and the term consanguinity is generally used when individuals are second cousins or closer.
However, there are other explanations for a relatively high level of identity by descent. For example, a high overall level of homozygosity can result from unusual recombination or segregation patterns during meiosis. It may also be observed for a distantly related couple who have multiple common ancestors. The latter circumstance may occur, for example, in individuals from an isolated population that arose recently from a small founding group or in populations where cousin marriages are common. For the above reasons, the SNP data themselves are not diagnostic of a specific degree of parental relatedness.7-8 Therefore, SNP data must be interpreted in the context of additional family and social history information and the clinician must determine whether it is appropriate to pursue the question of parental consanguinity for individual families.
Figure 2: Multiple AOH regions were identified, consistent with parental relatedness or identity by descent.

Autosomal Recessive Disease Risk
Regardless of whether AOH results from uniparental disomy (UPD) or identity by descent, homozygosity anywhere in the genome raises the possibility of recessive conditions.
Our reports alert the physician to the increased possibility of these conditions for regions of homozygosity greater than 10Mb. The referring physician can use this information in conjunction with clinical features and family history to determine whether mutation testing of individual genes is warranted.
Triploidy
Triploidy can be seen prenatally and appears at an appreciable frequency in miscarriages, but it is extremely rare postnatally.
The three types of triploidy are 69,XXX; 69,XXY; and 69,XYY. With aCGH, 69,XXY and 69,XYY triploidy can be detected, but not 69,XXX. The SNP Microarray allows for the detection of all types of triploidy due to the capability of detecting four genotypes (AAA, AAB, ABB, and BBB) rather than the normal three genotypes (AA, AB, and BB).
Genetic Counseling
A referral to a clinical genetics professional is often appropriate for individuals and families undergoing whole-genome microarray testing. This may be valuable both before and after testing.
Families should be aware of the possibility of a result of uncertain clinical significance and the need for parental blood samples to help interpret the change. Families should also understand that findings of AOH may require additional testing of the proband before a diagnosis can be made.
Clinical geneticists can guide testing strategies and further evaluate the patient in light of the test results. In some cases, it may be important to discuss the potential for discovery of parental consanguinity. Genetic counseling can also elicit a thorough family and social history, which can be critical in the interpretation of the SNP array results.
Limitations
This platform is not optimized to detect low-level mosaicism and uniparental disomy of the heterodisomy type.
This platform will not detect balanced alterations (reciprocal translocations, Robertsonian translocations, inversions, and balanced insertions), point mutations, or imbalances of regions not represented on the microarray.
The failure to detect evidence of uniparental disomy does not exclude the clinical diagnosis of an imprinting associated disorder. UPD may be of the heterodisomy type, which is not detected by the array, and mechanisms other than UPD can cause the disorder.9
Similar to the copy number-only CMA, the CMA copy number + SNP cannot detect balanced rearrangements and may not be capable of detecting low-level mosaicism. It also does not
detect point mutations, small deletions or insertions below the resolution of the assay, or other types of mutations, such as epigenetic changes.
Finally, test results are sometimes of uncertain clinical significance, and studies of additional family members may be required to assist with interpretation.
References
1. Miller DT et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749-764.
2. Shaffer LG et al. Working Group of the Laboratory Quality Assurance Committee of the American College of Medical Genetics. Genetics in Medicine. 2007;9:654-662.
3. Kearney HM et al. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genetics in Medicine. 2011;13(7);680-52.
4. Manning M et al. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genetics in Medicine. 2010;12:742-745.
5. Stavropoulos J and Shago M. CCMG guidelines for genomic microarray testing. Approved by the board of directors, Canadian College of Medical Geneticists. 2010.
6. SignatureChip OS Disorders Tested. Clinically recognized regions of the genome assayed by the SignatureChip OS(v4)/Signature PrenatalChip OS(v4). Signature Genomics from PerkinElmer, Spokane, WA 99207 USA. (www.signaturegenomics.com).
7. Kearney HM et al. Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations. Clin Lab Med. 2011;31(4):595-613.
8. Papenhausen P et al. UPD detection using homozygosity profiling with a SNP genotyping microarray. Am J Med Genet A. 2011;155A(4):757-68. ACMG guidelines.
9. Tucker, T et al. Uniparental disomy: can SNP array data be used for diagnosis? Genetics in Medicine. 2012;14:753-756.